publications
publications by categories in reversed chronological order.
2026
-
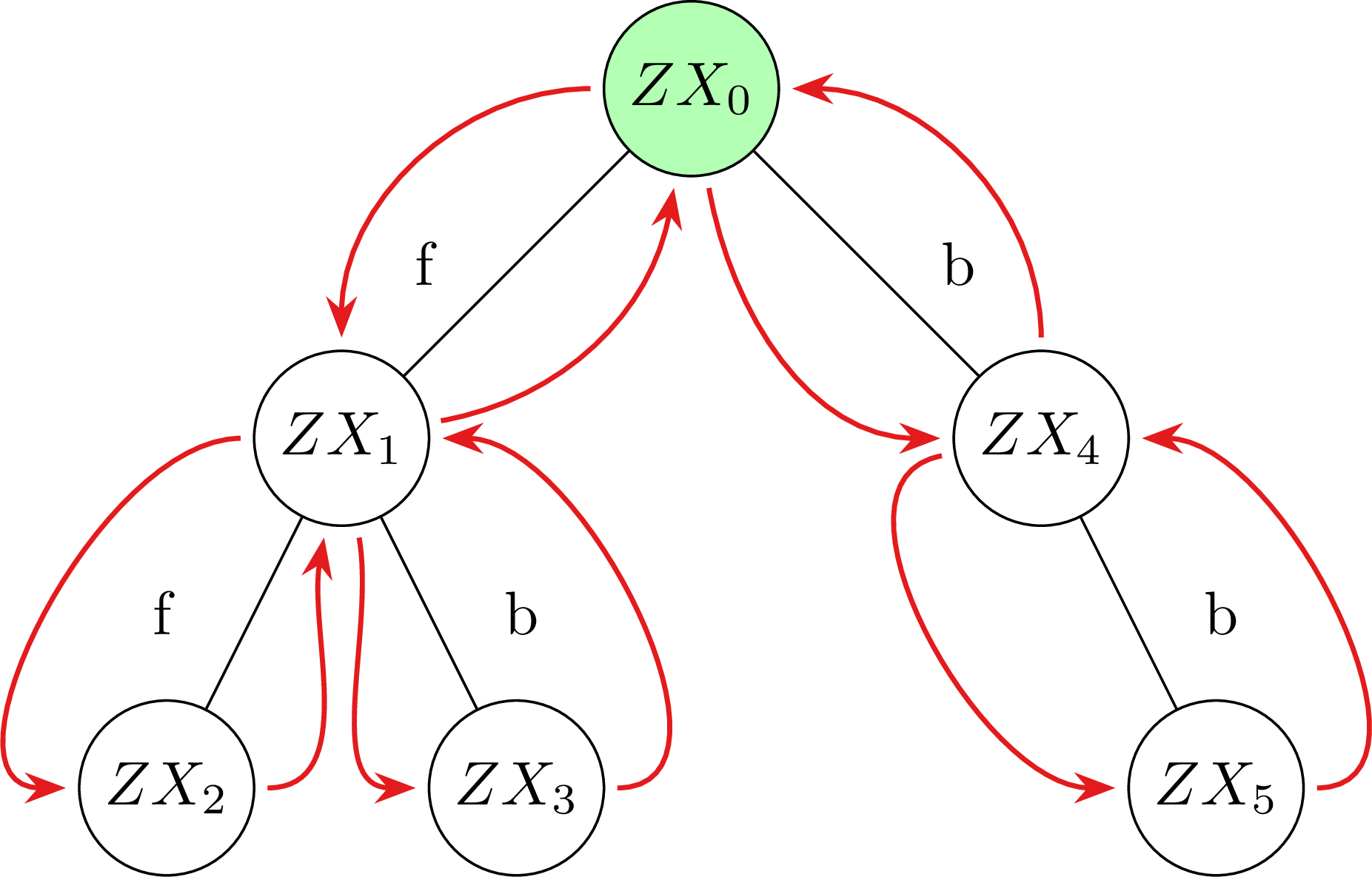 Exhaustive Search for Quantum Circuit Optimization Using ZX CalculusTobias Fischbach, Pierre Talbot, and Pascal BouvryConference paper for the International Conference on Optimization and Learning (OLA2025)
Exhaustive Search for Quantum Circuit Optimization Using ZX CalculusTobias Fischbach, Pierre Talbot, and Pascal BouvryConference paper for the International Conference on Optimization and Learning (OLA2025)Quantum computers allow a near-exponential speed-up for specific applications when compared to classical computers. Despite recent advances in the hardware of quantum computers, their practical usage is still severely limited due to a restricted number of available physical qubits and quantum gates, short coherence time, and high error rates. This paper lays the foundation towards a metric independent approach to quantum circuit optimization based on exhaustive search algorithms. This work uses depth-first search and iterative deepening depth-first search. We rely on ZX calculus to represent and optimize quantum circuits through the minimization of a given metric (e.g. the T-gate and edge count). ZX calculus formally guarantees that the semantics of the original circuit is preserved. As ZX calculus is a non-terminating rewriting system, we utilise a novel set of pruning rules to ensure termination while still obtaining high-quality solutions. We provide the first formalization of quantum circuit optimization using ZX calculus and exhaustive search. We extensively benchmark our approach on 100 standard quantum circuits. Finally, our implementation is integrated in the well-known libraries PyZX and Qiskit as a compiler pass to ensure applicability of our results.
2025
-
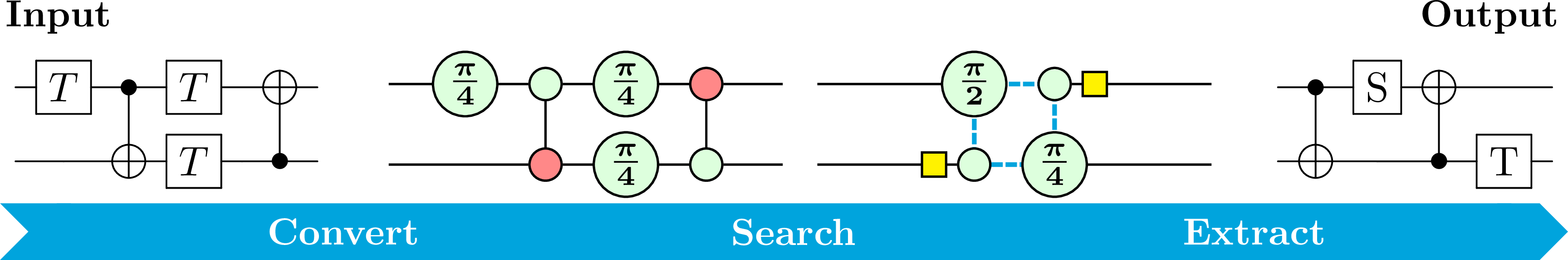 A Review on Quantum Circuit Optimization using ZX-CalculusTobias M. Fischbach, Pierre Talbot, and Pascal Bouvry
A Review on Quantum Circuit Optimization using ZX-CalculusTobias M. Fischbach, Pierre Talbot, and Pascal BouvryQuantum computing promises significant speed-ups for certain algorithms but the practical use of current noisy intermediate-scale quantum (NISQ) era computers remains limited by resources constraints (e.g., noise, qubits, gates, and circuit depth). Quantum circuit optimization is a key mitigation strategy. In this context, ZX-calculus has emerged as an alternative framework that allows for semantics-preserving quantum circuit optimization. We review ZX-based optimization of quantum circuits, categorizing them by optimization techniques, target metrics and intended quantum computing architecture. In addition, we outline critical challenges and future research directions, such as multi-objective optimization, scalable algorithms, and enhanced circuit extraction methods. This survey is valuable for researchers in both combinatorial optimization and quantum computing. For researchers in combinatorial optimization, we provide the background to understand a new challenging combinatorial problem: ZX-based quantum circuit optimization. For researchers in quantum computing, we classify and explain existing circuit optimization techniques.
-
 Exhaustive Search for Quantum Circuit Optimization using ZX CalculusTobias M. Fischbach, Pierre Talbot, and Pascal BouvryPoster presented at the EQAI 2025 Summer School, Lignano Sabbiadoro, Italy
Exhaustive Search for Quantum Circuit Optimization using ZX CalculusTobias M. Fischbach, Pierre Talbot, and Pascal BouvryPoster presented at the EQAI 2025 Summer School, Lignano Sabbiadoro, Italy -
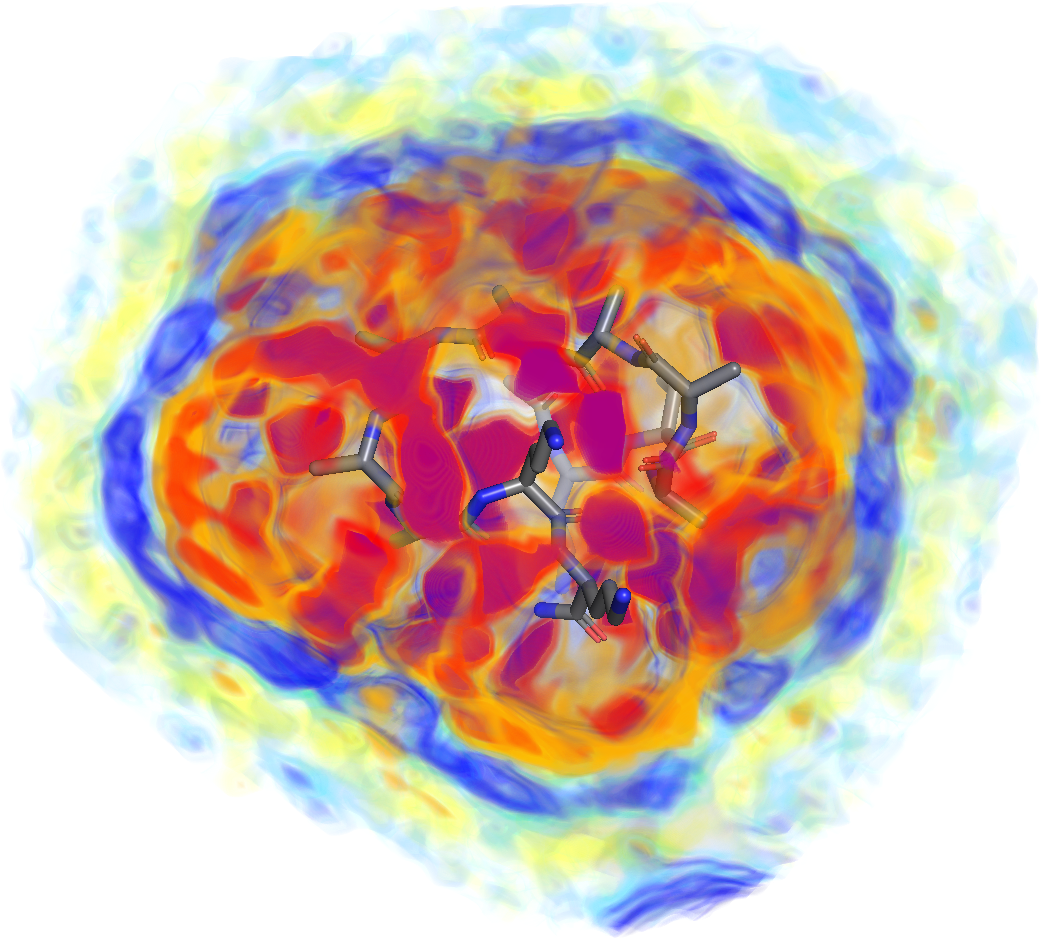 Hydration shells of globular and intrinsically disordered proteins: effects of amino acid composition, peptide conformation, and force fieldsJohanna-Barbara Linse, Tobias M. Fischbach, and Jochen S. Hub
Hydration shells of globular and intrinsically disordered proteins: effects of amino acid composition, peptide conformation, and force fieldsJohanna-Barbara Linse, Tobias M. Fischbach, and Jochen S. HubThe protein hydration shell is a key mediator of processes such as molecular recognition, protein folding, and proton transfer. How surface-exposed amino acids shape the hydration shell structure is not well understood. We combine molecular dynamics simulations with explicit-solvent predictions of small-angle X-ray scattering (SAXS) curves to quantify the contributions of all 21 proteinogenic amino acids to the hydration shell of the globular GB3 domain and the intrinsically disordered protein (IDP) XAO. We focus on two quantities encoded by SAXS curves: the hydration shell effect on the radius of gyration and the electron density contrast between protein and solvent. We derive an amino-acid-specific contrast score, revealing that acidic residues generate the strongest contrast with 1 to 1.5 excess water molecules relative to alanine, followed by cationic and polar residues. In contrast, apolar residues generate a water depletion layer. These trends are consistent across simulations with different water models. Around the XAO peptide, the hydration shell is generally far weaker compared to the globular GB3 domain, indicating unfavorable water–peptide packing at the IDP surface. The hydration shell effect on the radius of gyration of the IDP is strongly conformation-dependent. Together, the calculations show that the composition and spatial arrangement of surface-exposed amino acids govern the hydration shell structure, with implications for a wide range of biological functions and for hydration-sensitive experimental techniques such as solution scattering.Significance Hydration shells of biomolecules constitute a large fraction of the water in crowded cellular environments and play key roles in biological functions such as enzymatic reactions and conformational transitions. Small-angle X-ray scattering (SAXS) has shown that hydration shells differ in density from bulk water, yet how surface-exposed amino acids and protein surface geometry shape the hydration shell is not well understood. We combined molecular dynamics simulations with explicit-solvent SAXS predictions to quantify how surface-exposed chemical moieties and protein geometry drive variations in hydration shell density. Notably, the hydration shell of a globular protein differs markedly from that of an intrinsically disordered protein. Our study offers a comprehensive characterization of protein hydration and informs the interpretation of hydration-sensitive experimental techniques. Competing Interest Statement: The authors have declared no competing interest. Deutsche Forschungsgemeinschaft, https://ror.org/018mejw64, HU 1971/3-2, INST 256/539-1
- Exhaustive Search for Quantum Circuit Optimization using ZX CalculusTobias M. Fischbach, Pierre Talbot, and Pascal BouvryConference paper for the International Conference on Optimization and Learning (OLA2025)
Quantum computers allow a near-exponential speed-up for specific applications when compared to classical computers. Despite recent advances in the hardware of quantum computers, their practical usage is still severely limited due to a restricted number of available physical qubits and quantum gates, short coherence time, and high error rates. This paper lays the foundation towards a metric independent approach to quantum circuit optimization based on exhaustive search algorithms. This work uses depth-first search and iterative deepening depth-first search. We rely on ZX calculus to represent and optimize quantum circuits through the minimization of a given metric (e.g. the T-gate and edge count). ZX calculus formally guarantees that the semantics of the original circuit is preserved. As ZX calculus is a non-terminating rewriting system, we utilise a novel set of pruning rules to ensure termination while still obtaining high-quality solutions. We provide the first formalization of quantum circuit optimization using ZX calculus and exhaustive search. We extensively benchmark our approach on 100 standard quantum circuits. Finally, our implementation is integrated in the well-known libraries PyZX and Qiskit as a compiler pass to ensure applicability of our results.
- How protein hydration depends on amino acid composition, peptide conformation, and force fieldsJohanna-Barbara Linse, Tobias M. Fischbach, and Jochen S. Hub
The protein hydration shell is a key mediator of processes such as molecular recognition, protein folding, and proton transfer. How solvent-exposed amino acids shape the hydration shell structure is not well understood. We combine molecular dynamics simulations with explicit-solvent predictions of small-angle x-ray scattering (SAXS) curves to quantify the contributions of all 20 proteinogenic amino acids to the hydration shell of the globular GB3 domain and the intrinsically disordered protein (IDP) XAO. We focus on two quantities encoded by SAXS curves: the hydration shell effect on the radius of gyration and the electron density contrast between protein and solvent. We derive an amino acid-specific contrast score, revealing that acidic residues generate the strongest contrast with 1–1.5 excess water molecules relative to alanine, followed by cationic and polar residues. In contrast, apolar residues generate a water depletion layer. These trends are consistent across simulations with different water models. Around the XAO peptide, the hydration shell is generally far weaker compared with the globular GB3 domain, indicating unfavorable water-peptide packing at the IDP surface. The hydration shell effect on the radius of gyration of the IDP is strongly conformation-dependent. Together, the calculations show that the composition and spatial arrangement of solvent-exposed amino acids govern the hydration shell structure, with implications for a wide range of biological functions and for hydration-sensitive experimental techniques such as solution scattering.
2023
-
 Challenges in Automatic Software Optimization: the Energy Efficiency CaseTobias M. Fischbach, Emmanuel Kieffer, and Pascal BouvryConference paper for the International Conference on Optimization and Learning (OLA2023)
Challenges in Automatic Software Optimization: the Energy Efficiency CaseTobias M. Fischbach, Emmanuel Kieffer, and Pascal BouvryConference paper for the International Conference on Optimization and Learning (OLA2023)With the advent of the Exascale capability allowing supercomputers to perform at least 1018 IEEE 754 Double Precision (64 bits) operations per second, many concerns have been raised regarding the energy consumption of high-performance computing code. Recently, Frontier operated by the Oak Ridge National Laboratory, has become the first supercomputer to break the exascale barrier. In total, Frontier contains 9,408 CPUs, 37,632 GPUs, and 8,730,112 cores. This world-leading supercomputer consumes about 21 megawatts which is truly remarkable as Frontier was also ranked first on the Green500 list before being recently replaced. The previous top Green500 machine, MN-3 in Japan, provided 39.38 gigaflops per watt, while the Frontier delivered 62.68 gigaflops per watt. All these infrastructure and hardware improvements are just the tip of the Iceberg. Energy-aware code is now required to minimize the energy consumption of distributed and/or multi-threaded software. For example, the data movement bottleneck is responsible for 35-60% of a system’s energy consumption during intra-node communication. In an HPC environment, additional energy is consumed through inter-node communication. This position paper aims to introduce future research directions to enter now in the age of energy-aware software. The paper is organized as follows. First, we introduce related works regarding measurement and energy optimization. Then we propose to focus on the two different levels of granularity in energy optimization.
2022
-
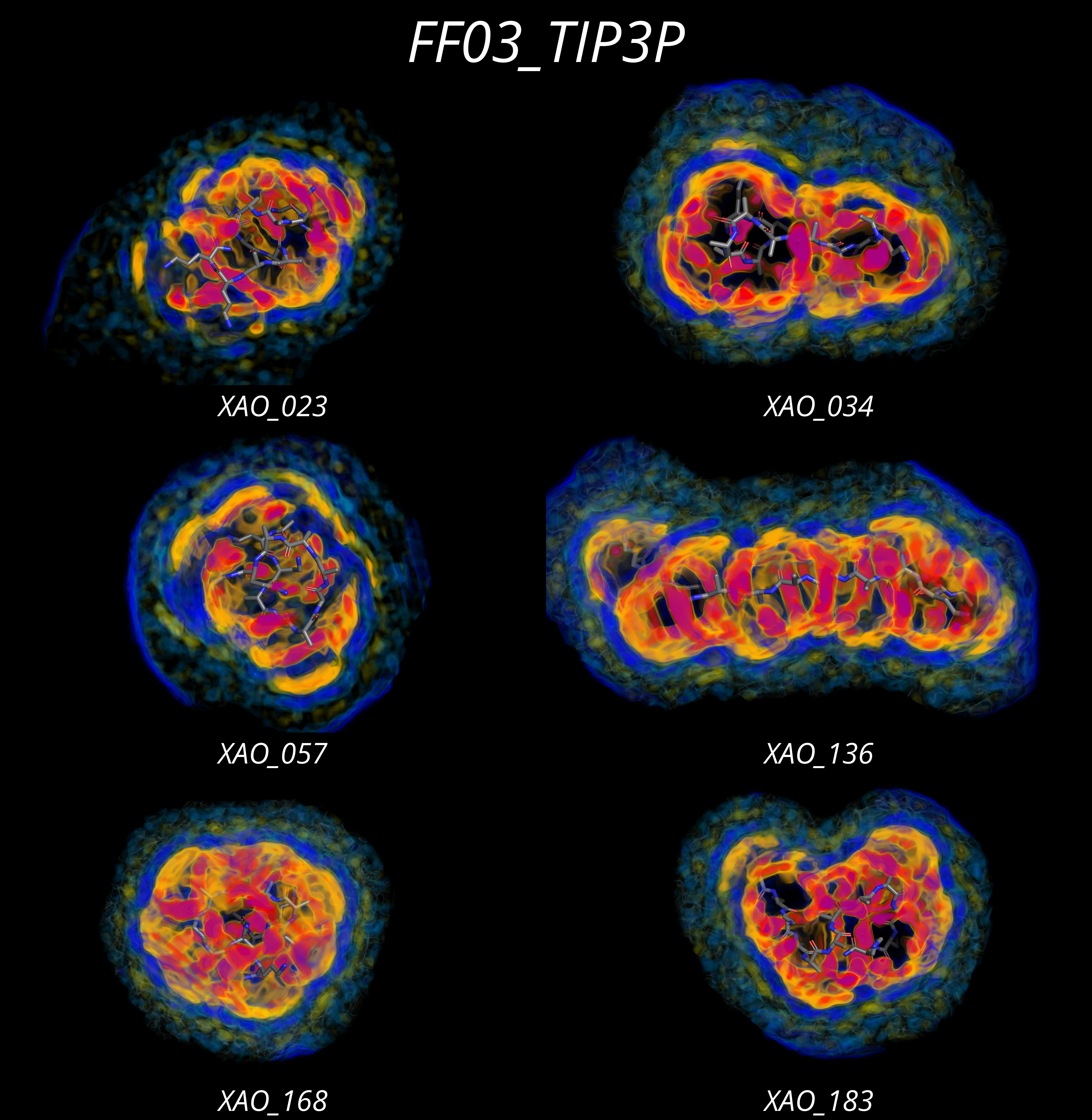 Hydration shell of intrinsically disordered proteinsTobias M. Fischbach and Jochen S. HubMaster thesis
Hydration shell of intrinsically disordered proteinsTobias M. Fischbach and Jochen S. HubMaster thesisThis work aims to leverage the power of molecular dynamics simulation to assess the hydration shell structure and its impact on the radius of gyration around intrinsically disordered proteins. Consequently, the long-term goal is to understand better the hydration shell and aid experimentalists in estimating the particle size in SAXS experiments more accurately. Proteins in a solution interact with the solvent, resulting in a surrounding hydration shell. On average, the hydration shell is denser compared to the water density of a pure solvent buffer. The hydration shell contrast denotes the difference in the water density of the solutions buffer and pure solvent buffer. It is a particular problem for SAXS experiments as they rely on the scattering intensity difference between the scattering intensity of the solution and solvent buffer to isolate the contribution of the protein to the scattering intensity. However, this is not entirely accurate as the final scattering intensity includes protein and hydration shell contrast contributions. In practice, this can lead to imprecise estimation of the radius of gyration through a Guinier analysis. Intrinsically disordered proteins lack a stable 3D structure and function as flexible linkers. Research interest in intrinsically disordered rapidly grows as it is more and more apparent that their lack of a stable structure allows them to perform their respective function. A scientific dispute whether XAO, an IDP, possesses a helix-like stable conformation or not leads to a well-measured NMR ensemble and an accurately determined radius of gyration through SAXS experiments. The scattering contribution of the hydration shell alone is commonly approximated with 10% of the pure solvent density. However, this approach neglects the hydration shell structure and density modulation from the protein water interaction. XAO is an artificial peptide that is intrinsically disordered and overall positively charged. We choose to study the hydration shell on this peptide because, due to a scientific dispute, whether a stable helix-like conformation exists leads to a well-measured NMR ensemble and precise SAXS experiments. Challenges encountered during this dispute were the correct conformational sampling of the forcefield and interpretation of the data. As the hypothesis about a helix-like stable structure of XAO highlights, force field parameters need to be chosen with care to reproduce experimental results. The simulation of intrinsically disordered proteins is challenging because IDPs do not possess a stable structural state; they need to be described by their conformational ensemble. However, comparing experimental and simulated ensembles reveals significant deviations for different force fields with two significant issues: (i) Incorrect secondary structure sampling (ii) Over-compact conformational ensemble The general approach of force fields to correct the secondary structure sampling is a refitting of backbone parameters in order to sample the dihedral angles correctly. All standard force fields produce an over-compact conformational ensemble of intrinsically disordered proteins. As the overall conformational size of intrinsically disordered proteins depends strongly on the protein water interaction, two specific hypotheses are currently investigated. Since introducing Small-angle X-ray scattering (SAXS), this low-resolution method has enjoyed increasing popularity. It is often used in combination with other experiments for structure determination to avoid overfitting. One reason for the success is that SAXS investigates the protein in solution. Hence, no crystallization artifacts are introduced, and the protein can adopt its natural folded state and investigate the size and shape of proteins in native conditions. From SAXS experiments, several different structural pieces of information can be obtained. The low scattering region of the excess intensity is characteristic of the particle size (Guinier region), and the radius of gyration can be extracted through a linear fit in a Guinier plot. Kratky plots permit the assessment of the flexibility of the protein and determine whether it is in a folded, partially unfolded, or disordered state. Finally, Porod-Debye plots contain volume information of the protein, while the pair distribution functions provide information about the longest diameter. Performing SAXS experiments on intrinsically disordered proteins imposes several challenges due to their high flexibility. It is not enough to take only one conformation into account compared to folded proteins. Instead, intrinsically disordered proteins are described by an ensemble of conformations. Therefore, NMR ensembles are combined with SAXS measurements. However, protein-water interaction leads to a density modulation surrounding the protein with an increased water density compared to the solvent-only buffer. Therefore, the excess intensity contains information about the protein and the hydration shell, propagating to the analysis. In order to clean the excess density from the hydration shell contribution, a hydration shell of 10% of the solvent density is commonly fitted. Nevertheless, this approach neglects the actual hydration shell structure. Therefore, this work introduces the notion of a hydration shell contrast between the shell and the average bulk water based on the solvent density included in the envelope. Combining the density difference with the corresponding volume results in the number of electrons contributing to the excess intensity. However, different water models and force fields cause a different shell structure. Therefore, this work aims to analyze the hydration shell and its contribution to the excess intensity and how the hydration shell structure impacts the radius of gyration compared to the traditional method of fitting the hydration shell with a fixed solvent density. We suspect, that the charged residues, as they are responsible for most hydrogen bonding, play a key role in the hydration shell structure. Furthermore, this work introduces an amino acid score. It describes the impact of the different amino acids on the hydration shell contrast.
2018
-
 Molekulardynamik Simulationen von Hydrophobin-DoppelschichtenTobias M. Fischbach and Jochen S. HubBachelor thesis
Molekulardynamik Simulationen von Hydrophobin-DoppelschichtenTobias M. Fischbach and Jochen S. HubBachelor thesis